Understanding Sickle Cell Disease: A Comprehensive Overview
Sickle Cell Disease (SCD) is a genetic disorder that affects millions of people worldwide, particularly those with African, Mediterranean, Middle Eastern, and South Asian ancestry. This complex condition has been the subject of extensive research, but there is still much to learn and understand about it. In this comprehensive overview, we will delve into the fundamental aspects of Sickle Cell Disease, from its genetic origins to its impact on individuals and potential treatment options.
I. Genetics of Sickle Cell Disease
Sickle Cell Disease is primarily a genetic disorder caused by a mutation in the HBB gene, which encodes a protein called hemoglobin. Hemoglobin is responsible for carrying oxygen throughout the body. In individuals with SCD, this mutation leads to the production of abnormal hemoglobin known as hemoglobin S (HbS).
II. Hemoglobin S and Red Blood Cells
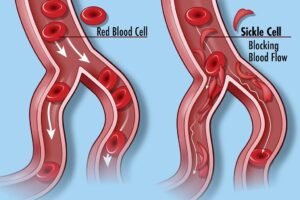
Hemoglobin S differs from normal hemoglobin (HbA) in its ability to change shape under certain conditions, particularly when exposed to low oxygen levels. This change in shape causes red blood cells to become rigid and assume a characteristic sickle or crescent shape. These abnormally shaped red blood cells are less flexible and can easily become trapped in blood vessels, leading to blockages and reduced oxygen delivery to tissues.
III. Clinical Manifestations of Sickle Cell Disease
Sickle Cell Disease presents a wide range of symptoms and complications, which can vary in severity among individuals. Common clinical manifestations include:
- Pain Crises: Patients with SCD often experience episodes of severe pain, known as pain crises, due to blocked blood flow.
- Anemia: The destruction of sickled red blood cells leads to anemia, causing fatigue and weakness.
- Organ Damage: Prolonged oxygen deprivation can damage vital organs, such as the heart, lungs, kidneys, and brain.
- Infections: SCD can weaken the immune system, making individuals more susceptible to infections.
IV. Diagnosis and Management
Diagnosing Sickle Cell Disease usually involves a blood test to identify the presence of abnormal hemoglobin. Once diagnosed, managing the condition becomes essential. Management strategies may include:
- Pain Management: Medications and lifestyle changes to alleviate pain during crises.
- Hydroxyurea: This medication can increase the production of a different type of hemoglobin (HbF) and reduce the frequency of painful crises.
- Blood Transfusions: Used to increase the number of healthy red blood cells in the body.
- Bone Marrow Transplantation: A potential cure for SCD, but it is a complex and risky procedure.
V. The Importance of Research and Awareness
While significant progress has been made in understanding and managing Sickle Cell Disease, there is still no universal cure. Continued research is crucial to developing more effective treatments and, ultimately, finding a cure. Raising awareness about SCD is equally important to promote early diagnosis, access to care, and support for affected individuals and their families.
Sickle Cell Disease is a complex genetic disorder that affects millions of people worldwide, particularly those of African descent. Understanding the genetic and physiological underpinnings of SCD is essential for both individuals living with the condition and the broader medical community. While there is no cure yet, advances in research and medical management offer hope for improving the quality of life for those affected by SCD. Increased awareness and support for research efforts can pave the way for a brighter future for individuals and families dealing with this challenging condition.
Comments